
Súbor:Phylogenetic tree of coronaviruses.jpg
Nie je dostupné vyššie rozlíšenie.
Phylogenetic_tree_of_coronaviruses.jpg (571 × 451 pixelov, veľkosť súboru: 75 KB, MIME typ: image/jpeg)
{kind=link}
Zhrnutie
| Popis |
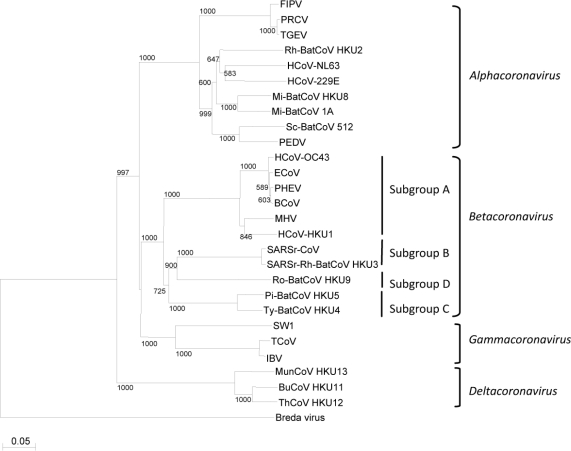
English: Phylogenetic analysis of RNA-dependent RNA polymerases (Pol) of coronaviruses with complete genome sequences available. The tree was constructed by the neighbor-joining method and rooted using Breda virus polyprotein. Bootstrap values were calculated from 1000 trees. 1118 amino acid positions in Pol were included. The scale bar indicates the estimated number of substitutions per 20 amino acids. All abbreviations for the coronaviruses were the same as those in Figure 1. |
| Dátum | |
| Zdroj | https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3185738/ |
| Autor | Patrick C. Y. Woo, Yi Huang, Susanna K. P. Lau, and Kwok-Yung Yuen |
Licencovanie
Tento súbor podlieha Creative Commons Attribution 3.0 Unported licencii
- Môžete slobodne:
- zdieľať – kopírovať, šíriť a prenášať dielo
- meniť ho – upravovať dielo
- Za nasledovných podmienok:
- uvedenie autorov – Musíte spomenúť autorov (jednotlivo alebo kolektívne), poskytnúť odkaz na licenciu a uviesť, či ste niečo zmenili. Môžete to urobiť ľubovoľným primeraným spôsobom, ale nie spôsobom naznačujúcim, že poskytovateľ licencie podporuje vás alebo vaše použitie diela.
História súboru
Po kliknutí na dátum/čas uvidíte ako súbor vyzeral vtedy.
| Dátum/Čas | Náhľad | Rozmery | Používateľ | Komentár | |
|---|---|---|---|---|---|
| aktuálna | 02:42, 7. marec 2020 | | 571 × 451 (75 KB) | Guest2625 | Uploaded a work by Patrick C. Y. Woo, Yi Huang, Susanna K. P. Lau, and Kwok-Yung Yuen from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3185738/ with UploadWizard |
Použitie súboru
Žiadne stránky neobsahujú odkazy na tento súbor.
Globálne využitie súborov
Nasledovné ďalšie wiki používajú tento súbor:
- Použitie na bn.wikipedia.org
- Použitie na en.wikipedia.org
- Použitie na fa.wikipedia.org
- Použitie na fr.wikipedia.org
- Použitie na ig.wikipedia.org
- Použitie na sq.wikipedia.org
- Použitie na su.wikipedia.org
{kind=link}